Способ лечения синдрома Fraley. Диагностика и методы лечения.
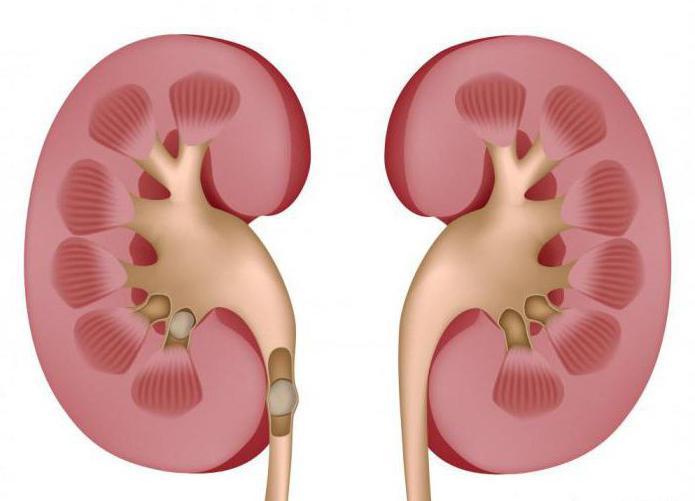
Синдром Фрейли получил свое название по имени американского уролога, описавшего врожденную аномалию сосудов в почках. Перекрещивание передних и задних ветвей сегментарной почечной артерии, в результате чего происходит сдавливание верхней лоханки либо лоханочно-мочеточникового сегмента. Вследствие этого аномального строения сосудов, синдром Фрейли характеризуется нарушением нормальных функций почки, что нередко влечет за собой образование песка и камней в почках, появлению признаков артериальной гипертензии, следов крови в моче. При наличии синдрома Фрейли часто возникают болевые ощущения в области поясницы, почечные колики.
Симптомы синдрома Фрейли
Симптомы синдрома Фрейли выражены появляющимися почечными коликами и болевыми ощущениями в пояснице. Иногда можно заметить следы крови в моче. Умеренная артериальная гипертензия также сопровождает клинические проявления синдрома Фрейли.
Лечение синдрома Фрейли
Лечение синдрома Фрейли проводится после проведения тщательного обследования пациента и постановки диагноза, который подтверждает или исключает наличие этой врожденной аномалии. В процессе обследования наши специалисты применяют самую современную методику и аппаратуру. Сообразно тяжести клинических проявлений заболевания, общего состояния пациента, его возраста и медицинских показаний может быть предложено хирургическое лечение. Оперативный метод являются единственным по-настоящему результативным при лечении синдрома Фрейли. Для получения лучших результатов опытный врач избирает наиболее эффективный метод хирургической операции. Внимательное отношение к каждому пациенту и высокий профессионализм наших врачей и медицинского персонала обеспечивают качественную медицинскую помощь и в операционный период и в процессе послеоперационного восстановления.
Некоторые врожденные почечные патологии не считаются болезнью, но их наличие может сопутствовать развитию заболеваний связанных с нарушением функционирования почек. Одной из таких аномалий развития является перекрещивание ветвей почечной артерии (Синдром Фрейли), которое сдавливает почечную лоханку сверху, мешая функционированию почек. По Международной классификации болезней (МК 10) он относится к XIV классу и имеет код 13 «Обстругивая уропатия и рефлюкс-уропатия».
Симптомы синдрома
Обычно синдром развивается в одной почке, но иногда аномалия сосудов наблюдается и в обеих почках. От этого зависит насколько ярко будут проявляться симптомы.
Врачи советуют обратить внимание на следующие признаки:
- Боли в области поясницы. Они проявляются со стороны больной почки, но в случае когда поражены обе боль носит опоясывающий характер.
- Частые почечные колики. Они свидетельствуют об наличие песка или камней в почках.
- Кровь в моче. Она может быть видна в моче, а может быть, обнаружена в результате анализов.
- Повышенное давление так называемая . Наличие синдрома Фрейли становится причиной высокого давления у детей и подростков.
- Уменьшение объема выделяемой мочи, вследствие чего наблюдается отечность.
У детей к симптомам может добавиться утомляемость, ухудшение памяти, сильная раздражительность.
Диагностика аномалии
Особенностью синдрома Фрейли, как и многих других врожденных почечных аномалий, является то, что он не имеет ярко выраженной симптоматики. Поэтому его трудно диагностировать, в большинстве случаев синдром Фрейли обнаруживают в результате обследования при обострении заболеваний мочевыводящей системы, беременности.
Выявить его при внешнем осмотре невозможно. Для постановки диагноза необходимы результата или рентгенографии, или УЗИ также обнаружить аномалию развития сосудов в почке можно и на МРТ. Но самым информативным считается метод почечной ангиографии, который позволяет точно определить наличие аномалии. Для диагностирования синдрома Фрейли у детей младше трех лет рекомендуется проводить доплерографию или МСКТ.
Для профилактики заболеваний и лечения почек наши читатели советуют Монастырский сбор отца Георгия. Он состоит из 16 полезных лекарственных трав, которые обладают крайне высокой эффективностью в очищении почек, в лечении почечных болезней, заболеваний мочевыводящих путей, а также при очищении организма в целом. Избавиться от боли в почках...»
Лечение синдрома
Лечению синдрома Фрейли зависит от степени патологии. Если затронута одна почка, а сосуды не сильно пережимают лоханку, то почки серьезно не страдают. При этом человек, может, даже не догадываться о наличии аномалии сосудов. В таких случаях лечение синдрома Фрейли не требуется, но при появлении симптомов необходимо лечения сопутствующих заболеваний и постоянное наблюдение.
Если же синдром Фрейли имеется на обеих почках, или происходит сильное сдавливание почечной лоханки, то, возможно, без оперативного лечения не обойтись. И хотя хирургический метод считается самым действенным, врач может его назначить только в самых серьезных случаях или при резком ухудшении состояния пациента.
Беременность и синдром Фрейли
Если у женщины был диагностирован синдром Фрейли, то она должна проинформировать об этом врача при постановке на учет по беременности. Так как наличие аномалии сосудов может привести к серьезным осложнениям во время вынашивания и родов. В идеале рекомендуется провести операцию по устранению порока развития, но делать это необходимо перед планированием беременности. Многие беременные даже не догадываются о наличие, пока его не обнаруживают на плановом обследовании.
Все беременные с синдромом Фрейли находятся под постоянным наблюдением, им проводят необходимое лечение. Особенно опасным считается второй триместр, когда осложнения проявляются особенно сильно. В некоторых случаях приходится прерывать беременность. Чаще всего после 30 недели, беременных кладут в стационар на сохранение. Обычно аномалия является показаниям к кесареву сечению.
Причины патологии
До сих пор врачи не могут точно сказать, что является причиной . Но в большинстве своем они выделяют несколько причин:
- Генетические и наследственные.
- Курение и употребление алкоголя во время беременности, зачатия.
- Инфекционные, хронические заболевания беременных.
- Прием определенных лекарств в период беременности.
- Экологические факторы.
Конечно, синдром Фрейли может приносить немало проблем, но не стоит паниковать если его диагностировали. Ведь с такой аномалией даже берут служить в армию. И если посещать для профилактического осмотра врача, выполнять его рекомендации, стараться придерживаться здорового образа жизни, то он не беспокоить да конца жизни.
И немного о секретах...
Вы когда-нибудь мучались от проблем из-за боли в почках? Судя по тому, что вы читаете эту статью - победа была не на вашей стороне. И конечно вы не по наслышке знаете что такое:
- Дискомфорт и боль в пояснице
- Утренние отеки лица и век отнюдь не добавляют Вам уверенности в себе...
- Как-то даже стыдно, особенно если Вы страдаете частым мочеиспусканием...
- К тому же, постоянная слабость и недомогания уже прочно вошли в Вашу жизнь...
Синдром Фрейли - это патология развития кровеносных сосудов в почках, когда почечные лоханки с одной или двух сторон передавливаются. Считается врожденной из-за возникновения еще в утробе матери. Это не заболевание, но при этом имеются симптомы сопутствующих болезней, при которых нарушается функционирование одной или обеих почек.
Описание синдрома Фрейли было дано еще в 1966 году. Произвел его американский уролог, в честь которого он и назван. Синдром может иметь как левостороннюю, так и правостороннюю локализацию. В основном патология захватывает только одну почку, но бывает и двойное поражение. Обе почечные лоханки расширяются, поскольку нарушается проходимость мочеточника.
Факторы риска
Имеются определенные факторы, которые могут спровоцировать неправильное формирование или остановку развития почек. В этой связи появляются аномалии в сосудах, которые снабжают орган кровью.
Произойти это может из-за:
- наследственности;
- различных генетических аномалий;
- неблагоприятных факторов, воздействующих на плод во время беременности и негативно сказывающихся на его развитии;
- острых и хронических заболеваний у матери;
- приема лекарственных препаратов во время вынашивания ребенка;
- вредных привычек: пристрастия к спиртному, употребления наркотических средств, курения;
- влияния некоторых экологических и физических факторов на беременную женщину и развитие плода: высоких показателей температуры, длительного нахождения в экологически неблагоприятных условиях, вредных условий труда, воздействия ионизирующего излучения.
В результате возникает синдром Фрейли. Лечение симптоматическое не всегда помогает. Только при помощи оперативного вмешательства можно помочь пациенту.
Симптомы синдрома Фрейли
Сосуды перекрещиваются и передавливают верхнюю часть почки. В результате появляется определенная симптоматика, которая указывает на развитие патологии в почках:
Боль в поясничном отделе с одной или двух сторон;
Острая боль в области почки (почечная колика) ;
Кровь в моче;
Умеренное повышение давления;
Уменьшение выделяемого объема мочи.
Осложнения и последствия
Синдром Фрейли не является опасным для жизни. Но он вызывает патологические процессы в почках, поэтому не исключено развитие следующих осложнений:
- Утомляемость.
- Раздражительность.
- Ухудшение внимания и памяти.
- Мочекаменная болезнь.
- Пиелонефрит.
- Почечная недостаточность.
Диагностика синдрома Фрейли
В ходе диагностики врачам необходимо не только выявить патологию почек, но и обнаружить ее причину, а именно наличие у человека синдрома Фрейли. В этой связи определяется степень нарушения работы этого важного парного органа, а также необходимость в хирургической операции. Это поможет предотвратить дальнейшую компрессию лоханок почек сосудами.
Собирается анамнез у взрослого человека или у родителей малыша, если проявляется синдром Фрейли у детей. Но это не раскрывает полную картину болезни. Проводится общий анализ крови, мочи, микрофлоры для выявления воспалительного процесса в организме и присутствия песка или камней. Более трудно определение места локализации воспаления и образования камней, а также выявление причины (то есть наличия синдрома Фрейли и, как результат, сдавливания почек аномальными сосудами).
Лечение синдрома Фрейли

Как лечится синдром Фрейли? Это частый вопрос.
На основании результатов диагностики назначается терапия. В зависимости от степени компрессии почечных лоханок, а также от сопутствующих заболеваний она может быть различной. Если лоханка пережимается сосудами не сильно, то врожденная патология никак себя не проявит. До первых признаков не потребуется лечить синдром Фрейли. Симптомы могут так и не возникнуть.
При значительных нарушениях уродинамики должны быть приняты меры для избавления от симптоматики. Однако это не решит проблему синдрома Фрейли. Сдавливание лоханок будет мешать дальнейшему нормальному функционированию органа. Пиелонефрит станет хроническим, наличие камней - постоянным, давление - все время повышенным.
Поэтому помочь сможет только операция, если, конечно, позволяет возраст и здоровье больного.
Оперативное лечение
Различными методами хирургического лечения синдрома Фрейли являются сосудистые либо операции на верхних мочевыводящих путях. К ним относится:
- Инфундибулопластика (чашечно-лоханочного соустье увеличивается в размерах).
- Инфундибулоанастомоз (хирургически перемещается сосуд, затем фиксируется при помощи анастомоза).
- Инфундибулопиелонеостомия (перемещается сосуд в искусственное русло между чашкой и лоханкой).
- Каликопиелонеостомия (является идентичной вышеназванной).
Только врач сможет определить, какая именно нужна пациенту операция. Синдром Фрейли может проявляться с разной степенью тяжести.
Все эти методы могут вызвать различные осложнения:
- Образование мочевых затеков.
- Воспаление сосудистой ножки почки (педункулит).
- Образование грубых рубцов, приводящих к стенозу сосудов.
Интрарентальная вазопексия является новым хирургическим методом в лечении синдрома Фрейли. Она заключается в том, что производится разобщение мочевых путей и патологически расположенного сосуда. Это не приводит к вскрытию мочевых путей, значит, осложнений не будет. Особенно если обнаружен синдром Фрейли у детей.
Консервативное лечение синдрома Фрейли
В качестве консервативного лечения эффективным будет назначение: «Каптоприла», «Эналаприла», «Даприла», «Фоззиноприла», «Каптопреса», «Рениприла», «Энапа» и других медикаментов, относящихся к разряду ингибиторов АПФ.
При наличии боли в области почек показан прием спазмолитиков (снимают спазм и купируют болевые ощущения) и комбинированных обезболивающих средств: «Спазмила», «Спазмалгона», «Ависана», «Но-Шпы», «Папаверина», «Баралгина», «Новигана», «Спазмалина», «Бралангина», «Реналгана», «Платифиллина» и других.
Часто эффективным может оказаться физиотерапевтическое и гомеопатическое лечение. Народные средства для лечения почечной патологии тоже могут помочь.
Ведь очень неприятно, когда болят почки. Синдром Фрейли эффективно лечится. Поэтому нужно использовать все средства.
Синдром Фрейли: прогноз

Прогноз синдрома Фрейли зависит от того, насколько поражена почка. Следует для скорейшего выздоровления своевременно обратиться за помощью в медицинское учреждение. Лечение должно быть комплексным. Необходимость в терапии и вовсе может отсутствовать. А некоторые пациенты не решаются на операцию и мучаются на протяжении долгих лет. А зря, поскольку после хирургического вмешательства прогноз является самым благоприятным.
Мы подробно рассмотрели синдром Фрейли, симптомы и лечение описаны.
Согласно данным статистики, свыше 35% врожденных пороков связано с нарушением работы мочевыводящих путей. Одним из таких заболеваний является синдром Фрейли, который получил свое название по имени врача-уролога из США, впервые описавшего его. Как правило, такого рода аномалии определяются при беременности женщины или в подростковом возрасте. Чаще всего на протяжении длительного срока болезнь протекает незаметно.
Наследственная болезнь почек, которая затрагивает работу мочевыводящих путей имеет название - «синдром Фрейли».
Понятие и симптоматика
Синдром Фрейли - это врожденная почечная аномалия, образующаяся путем перекрещивания передних и задних ветвей почечной артерии и сдавливания лоханочно-мочеточникового сегмента либо верхней части лоханки. В результате почки перестают функционировать в нормальном режиме. Из-за аномалии в строении сосудов нарушается их работа, что в дальнейшем приводит к следующим последствиям:
 Синдром Фрейли не редко проявляется появлением крови в моче, коликами и образованием камней с песком в почках.
Синдром Фрейли не редко проявляется появлением крови в моче, коликами и образованием камней с песком в почках. - образованию в почках камней и песка;
- возникновению признаков артериальной умеренной гипертензии;
- появлению следов крови в моче (макро- и микрогематурии);
- болевым ощущениям в поясничной области;
- коликам в почках (обусловлено вторичным нефролитиазом- почечнокаменной болезнью).
Обнаружить симтоматику заболевания можно и в ходе эмбрионального развития почечной сосудистой системы. При этом структура органов сохраняется, однако есть вероятность остановки их развития. Обычно синдром задевает одну почку (может образоваться слева или справа). В некоторых случаях возможно нарушение работы обеих почек. Из-за препятствия для оттока мочи синдром может сопровождаться расширением лоханки (справа или слева), расширением почечных чашечек.
При обнаружении заболевания важно, чтобы период беременности и рождение ребенка проходили под наблюдением лечащего врача. Женщинам с синдромом Фрейли или другими врожденными почечными пороками только после операции разрешается вынашивание плода. Причиной является повышенное артериальное давление, которое преследует больного. Без операционного вмешательства беременность протекает тяжело. Бывают случаи, когда приходится прерывать ее на сроках после 4-х месяцев. Если операция прошла положительно, функции почек были восстановлены, в период беременности необходимо предупредить о перенесенном вмешательстве своего лечащего врача-гинеколога. На протяжении всего периода вынашивания плода женщине нужно проходить обследования, сдавать назначенные доктором анализы, консультироваться у нефролога.
 Беременность при синдроме Фрейли может быть усложнена для роженицы, но ребёнку не угрожает сей недуг.
Беременность при синдроме Фрейли может быть усложнена для роженицы, но ребёнку не угрожает сей недуг.
В некоторых случаях возможна и госпитализация, беременная женщина будет наблюдаться у врачей в стационарных условиях. Довольно часто заболевание почек обостряется на 15−16- недельном или 26−30- недельном сроке. Определить обострение синдрома можно по следующим признакам:
- сильно отекают конечности;
- задерживается отделение мочи;
- во время выведения мочи из организма отмечаются дискомфортные ощущения и даже боли.
На сроке от 30-ти недель причиной осложнения является быстрый рост матки, давящей на мочеточники. При проявлении вышеуказанных признаков беременную женщину, страдающую синдромом Фрейли, нужно срочно госпитализировать. Врожденные почечные пороки часто не обходятся без кесарева сечения. Важно отметить, что при этом практически нет угрозы для ребенка. Для рожающих женщин с аномалиями развития почек организованы специализированные роддома, где в обязательном порядке среди штатных сотрудников есть врач-нефролог и уролог. Сразу после рождения ребенка врачи проводят комплексное обследование младенца. Хотя аномалия почек и является врожденным заболеванием, современная медицина способна вылечить синдром Фрейли. После операционного вмешательства пациенты возвращаются к обычной жизни.
Лечение. Проводится консервативное лечение, направленное на борьбу с инфекцией мочевой системы. При хирургических осложнениях дистопии почки (гидронефроз, мочекаменная болезнь и т.д.), а также болевом синдроме применяют оперативное лечение. Ввиду морфофункциональной незрелости органа показания к нефрэктомии расширены. Однако, при сохранении функции применяют корригирующие органосохраняющие вмешательства с фиксацией почки в физиологически выгодном положении.
Нефроптоз характеризуется смещением почки в вертикальном положении ребенка на 1,5 и более поясничных позвонков, при этом могут возникать перегибы мочеточника, пиело- и каликоэктазия в результате уродинамических нарушений.
В последнее время нефроптоз относят к висцеральным проявлениям дисплазии соединительной ткани. Клинически заболевание наиболее часто проявляется в старшем возрасте в виде болевого абдоминального синдрома. Характерны боли в поясничной области, они провоцируются физической нагрузкой, подъемами тяжести, прыжками. Иногда отмечаются симптомы артериальной гипертензии, изолированный мочевой синдром (ортостатическая протеинурия, микрогематурия, лейкоцитурия). В связи с нарушением уродинамики может присоединяться хронический пиелонефрит. Диагноз верифицируется при проведении ультразвукового исследования почек и экскреторной урографии в горизонтальном и вертикальном положении больного. Лечение зависит от стадии заболевания и наличия осложнений. В начальных стадиях нефроптоза, когда имеются невыраженные клинические проявления и отсутствуют нарушения гемо- и уродинамики, проводится консервативное лечение. Консервативное лечение включает лечебную физкультуру
Аномалии формы обозначаются как сращенные почки. К ним относят подковообразную, галетообразную, L-образную, S-образную, I-образную, опухолевидную и другие более редкие формы сращений. Наиболее часто встречается подковообразная почка, при этом почки срастаются преимущественно верхними полюсами. Аномалии формы нередко сочетаются с дистопией, ротацией, дисплазией. Клинически могут проявляться болевым абдоминальным синдромом, дизурией, пальпируемым опухолевидным образованием, гипертензией, нередко присоединяется пиелонефрит, гидронефроз. Диагноз выставляется при проведении ультразвукового исследования почек и экскреторной урографии.
Лечение. Показанием к оперативному лечению являются: выраженные боли в животе, мочекаменная болезнь, гидронефроз, рецидивирующий вторичный пиелонефрит. При сохраненной функциональной способности обеих половин производят рассечение перешейка (истмотомия). В случаях гидронефроза эту операцию сочетают с пластикой пиелоуретерального сегмента. Утративший функцию патологический сегмент подлежит удалению (геминефрэктомия). При отсутствии показаний к оперативному лечению, проводится консервативная терапия, включающая прежде всего, антибактериальную терапию хронического обструктивного пиелонефрита.
В группе анатомических аномалий органов мочевой системы выделяют врожденные обструктивные нефропатии, общим признаком которых является нарушение оттока мочи, повышение внутрилоханочного давления и, как следствие, расширение чашечно-лоханочной системы, ведущее к атрофии почечной паренхимы. К ним относят: первичный пузырно-мочеточниковый рефлюкс, синдром Фрейли (сдавление верхней чашечки аномальным артериальным сосудом), гидронефроз (стеноз пиелоуретерального сегмента), мегауретер (стеноз в терминальном отделе мочеточника) и инфравезикальная обструкция. Наиболее часто у детей встречаются пузырно-мочеточниковый рефлюкс, синдром Фрейли и врожденный гидронефроз.
Синдром Фрейли – врожденный изолированный гидрокаликоз в результате сдавления шейки верхней группы чашечек внутриорганным аномалийно расположенным сосудом.
Аномалия проявляется болями в поясничной области, микрогематурией, симптомами пиелонефрита. При проведении ультразвукового исследования почек, выявляется изолированная каликоэктазия верхней группы чашек. На экскреторных урограммах помимо изолированного гидрокаликоза выявляется линейный дефект наполнения в проекции аномального сосуда, проходящего в области шейки верхней группы чашек. Более достоверно диагноз устанавливается при проведении почечной ангиографии. Возможен изолированный гидрокаликоз в результате сдавления шейки нижней группы чашечек аномально расположенным сосудом – синдром «нижней чашечки». Лечение консервативное. В редких случаях показанием к оперативному вмешательству может быть выраженный болевой синдром или упорный пиелонефрит. Однако реконструктивное вмешательство, как правило, не удается, и проводят резекцию верхнего полюса пораженной почки.
Врожденный гидронефроз – стойкое прогрессирующее расширение почечной лоханки и чашечек в результате нарушения оттока мочи в пиелоуретеральном сегменте вследствие его обструкции.
Наиболее частыми причинами этой обструкции являются: стенозы мочеточника, сдавление его добавочными или аномально расположенными сосудами, высокое отхождение мочеточника от лоханки, фиксированный перегиб мочеточника, клапаны мочеточника, сдавление мочеточника фиброзными тяжами. Обструкция мочеточника в пиелоуретеральном сегменте вызывает повышение внутрилоханочного давления, прогрессирующую дилятацию чашечно-лоханочной системы, атрофию почки, прогрессирующее ухудшение функции почки и ее гемодинамики, а также развитие хронического пиелонефрита. Выделяют 4 степени гидронефроза: I степень – прегидронефроз, пиелоэктазия или интермиттирующий гидронефроз, II и III степень – собственно гидронефроз с большим или меньшим сохранением функции почки, IV степень – гидронефроз с резким истончением паренхимы и полной потерей функции органа. Клинически гидронефроз проявляется болями в животе, в поясничной области, рвотой, артериальной гипертензией, при пальпации живота может определяться опухолевидное образование в брюшной полости. Нередко гидронефроз осложняется присоединением хронического вторичного обструктивного пиелонефрита. При ультразвуковом исследовании выявляют увеличение размеров почки, расширение чашечно-лоханочной системы, истончение коры. При проведении экскреторной урографии выявляются замедление появления контраста на стороне поражения; увеличение лоханки и, особенно, чашечек («монетовидные» чашечки); резкое нарушение или отсутствие оттока мочи из лоханки при стандартном исследовании (необходимо проведение отсроченных рентгенограмм через 1, 2 и 4 часа). На радиоизотопной ренографии характерен обструктивный тип кривой. Ретроградная пиелография (стеноз, перегиб пиелоуретерального сегмента), ангиография (добавочный сосуд и особенности кровоснабжения почек) позволяют уточнить причину гидронефроза и характер предполагаемой хирургической коррекции.
Лечение. Консервативное лечение первой стадии заболевания направлено на стимуляцию оттока мочи, лечение пиелонефрита. Кроме антибактериальных средств применяют физиотерапию, лечебную гимнастику, массаж. При отсутствии эффекта проводят хирургическое лечение. В первых трех стадиях заболевания выполняют органосохраняющие операции, направленные на восстановление проходимости пиелоуретерального сегмента (пластика прилоханочного отдела мочеточника). В последней стадии показана нефрэктомия. В далеко зашедших стадиях двустороннего гидронефроза или при поражении единственной почки применяют двухэтапное оперативное лечение. На первом этапе некладывают разгрузочную нефростому, а затем, в зависимости от функционального состояния почек, выполняют пластику пиелоуретерального сегмента или нефрэктомию.
АНОМАЛИИ ФОРМИРОВАНИЯ ПОЧЕЧНОЙ ТКАНИ С ДЕФИЦИТОМ ПАРЕНХИМЫ
Аномалии формирования почечной ткани с дефицитом паренхимы включают гипоплазии почек.
Гипоплазия почек – врожденное уменьшение органа в размерах, превышающих 2 сигмальных отклонения от нормы. Выделяют нормонефроническую (простую) и олигомеганефроническую гипоплазию почек.
При нормонефронической (простой) гипоплазии отмечается уменьшение размеров (массы) почки при сохранении гистологического строения, то есть имеется нормальное количество неизмененных в размерах и строении нефронов, уменьшено количество чашечек до 5 или менее и имеется гипоплазия почечной артерии. Клинически данная аномалия ничем не проявляется, выявляется случайно при ультразвуковом исследовании почек. Иногда возникает необходимость проведения дифференциального диагноза с вторично сморщенной почкой, рефлюкс-нефропатией. С этой целью проводится функциональное исследование почек, экскреторная урография, микционная цистография, допплерография почечных сосудов, при необходимости – нефробиопсия с гистологическим, электронномикроскопическим и иммунногистохимическим исследованиями. Прогноз при односторонней гипоплазии благоприятный.
Лечение. При односторонней гипоплазии при отсутствии клиники, лечение не требуется. При осложнении пиелонефритом или нефрогенной гипертензией показано соответствующее лечение, в последнем случае – вплоть до нефрэктомии гипоплазированной почки. При двусторонней гипоплазии довольно быстро наступает ХПН, время появления которой зависит от выраженности порока. При резкой двусторонней гипоплазии почек дети погибают в раннем возрасте.
Олигомеганефроническая гипоплазия – всегда двусторонняя гипоплазия с самопроизвольным уменьшением числа функционирующих нефронов и развитием хронической почечной недостаточности. При данной аномалии уменьшено число нефронов, увеличены их размеры в 2-2,5 раза, увеличен их объем в 7-10 раз, в последующем развивается прогрессирующий склероз почек. Клиника появляется на первом году жизни ребенка в виде рвоты, признаков дегидратации организма, лихорадки, снижения массы тела. После года у ребенка отмечается артериальная гипертензия, полиурия, полидипсия, протеинурия, гематурия, в дальнейшем происходит развитие хронической почечной недостаточности. Прогноз при олигомеганефронической гипоплазии неблагоприятный. Лечение симптоматическое с последующим проведением программного гемодиализа и трансплантации почки.
АНОМАЛИИ ДИФФЕРЕНЦИРОВКИ (СТРУКТУРЫ) ПОЧЕК
Аномалии дифференцировки (структуры) почек включают дисплазии почек и поликистоз.
Дисплазии почек - аномалии, в основе которых лежит изменение первичной структуры почечной ткани с сохранением эмбриональных образований: примитивных протоков и канальцев, очагов недифференцированной мезенхимы.
Морфологическим субстратом является наличие очагов мезенхимы, примитивных клубочков, канальцев и протоков. Выделяют безкистозные и кистозные дисплазии. Для последних характерно наличие в паренхиме почек кист, разделенных самой почечной тканью или соединительнотканными прослойками. В зависимости от распространенности различают тотальную, очаговую и сегментарные формы кистозной дисплазии. При тотальной форме выделяют апластический, гипопластический, гиперпластический и мультикистозный варианты. Клинически дисплазии, как правило, протекают бессимптомно и проявляются изолированным мочевым синдромом в виде микрогематурии, протеинурии или их сочетаний. К кистозным дисплазиям относят:
Медуллярную кистозную болезнь (нефронофтиз Фанкони), для которой характерно диспластически-дегенеративное поражение преимущественно мозгового вещества почек, обилие кист из собирательных трубочек, гиалинизация и склерозирование клубочков и интерстиция, лимфоцитарная инфильтрация стромы.
Почки уменьшены в размерах, часто сохраняют эмбриональную дольчатость. Клинически болезнь чаще проявляется к 5-6 годам в виде полиурии, полидипсии, гипоизостенурии, отставании в физической развитии и постепенно прогрессирующей почечной недостаточности. Нефрофтиз Фанкони часто но& #1089;ит семейный характер с аутосомно-рецессивным и реже доминантным типом наследования. Лечение симптоматическое, при развитии хронической почечной недостаточности – гемодиализ и трансплантация почки. Мультикистозная почка представляет собой конгломерат различного размера тонкостенных кист, между которыми практически отсутствует почечная паренхима и лоханка. Процесс, как правило, односторонний.
Поликистоз почек. В основе заболевания лежит нарушение структур органа в результате превращения части паренхимы в кисты различного диаметра.
Проявляется в 2 формах: первая - преимущественно у новорожденных и детей раннего возраста с аутосомно-рецессивным типом наследования, вторая - у взрослых с аутосомно-доминантным типом наследования (взрослый тип). Поликистоз взрослого типа обусловлен неправильным соединением прямых и извитых канальцев в период внутриутробного развития, что ведет к нарушению оттока первичной мочи из проксимальных отделов нефрона, расширению слепо заканчивающихся канальцев и образованию из них кист. В процессе образования кист участвуют 3 типа нарушений: 1) изменение компонентов базальной мембраны; 2) нарушение пролиферации эпителия кист; 3) нарушение развития и поляризации эпителия кист. Морфологически почки всегда увеличены, наружная поверхность изменена из-за выступающих из-под капсулы кист. На разрезе почки определяются различной величины кисты в мозговом и корковых слоях, заполненные жидким или желеобразным содержимым. Состояние кист неоднородно, они могут формироваться из любых отделов нефрона. Клинические признаки поликистоза чаще выявляются через 10-20 лет. У детей заболевание протекает бессимптомно или регистрируется болевой или/и мочевой синдром в виде гематурии. Нередко выявляется поликистоз печени, аневризма мозговых сосудов, коарктация аорты, врожденные дефекты глаз. Лечение консервативное, при нарастании почечной недостаточности показан хронический гемодиализ с последующей трансплантацией. При поликистозе детского типа в отличие от взрослого типа почки сохраняют форму и гладкую поверхность, кисты представляют собой расширение дистальных канальцев, которые выстланы недифференцированным кубической или цилиндрической формы эпителием, кора не подвергается обширному повреждению, встречаются больше участков неповрежденной паренхимы. Практически у всех больных определяются множественные кисты в печени с пролиферацией желчных протоков и фиброзом. Клиническая картина зависит от степени кистозного перерождения почек и печени. Для новорожденных в типичных случаях характерно лицо Поттера (уплощенный нос, западающий подбородок, эпикант, гипертелоризм, микрогнатия), аномалии конечностей, спонтанный пневмоторакс, дыхательная недостаточность. Брюшная стенка растянута, нередко в форме «сливого живота», легко пальпируются увеличенные почки. Возможна анурия и почечная недостаточность, гематурия и артериальная гипертензия. Поликистоз часто осложняется вторичным пиелонефритом с торпидным течением. Смерть от прогрессирующей почечной недостаточности может наступить в возрасте от нескольких недель до нескольких месяцев. У подростков основную проблему представляет портальная гипертензия и рецидивирующая гематурия. Ведущим методом диагностики поликистоза является эхография в антенатальный (с 20 недели) и постнатальный периоды. Лечение паллиативное.
Наследственный нефрит
Наследственный нефрит – генетически детерминированное неиммунное нефритоподобное заболевание, проявляющееся гематурией и (или) протеинурией и часто сочетающееся с патологией слуха и реже зрения.
Заболевание передается по аутосомно-доминантному типу, сцепленному с Х-хромосомой (80-85%), аутосомно-рецессивному или аутосомно-доминантному типу наследования. Генные мутации приводят к нарушению трехспиральной структуры коллагена (альфа цепи коллагена 4 типа), что вызывает изменение не только базальных мембран почки, но и аналогичных структур уха и глаза. Выделяют 3 варианта наследственного нефрита.
Синдром Альпорта, для которого характерно наследственный нефрит с гематурией, тугоухость и поражение глаз. Заболевание наследуется по доминантному, сцепленному с Х-хромосомой, типу наследования. Течение нефрита прогрессирующее с исходом в хроническую почечную недостаточность.
Наследственный нефрит без тугоухости, характеризующий прогрессирующим течением с исходом в хроническую почечную недостаточность. Заболевание наследуется по доминантному, сцепленному с Х-хромосомой, типу наследования.
Семейная доброкачественная гематурия, которая протекает доброкачественно с благоприятным прогнозом. Заболевание наследуется по аутосомно-доминантному или аутосомно-рецессивному типу наследования. При аутосомно-доминантном типе наследования отмечается тромбоцитопения.
При морфологическом исследовании определяются диспластические, дистрофические, пролиферативные изменения, фокально-сегментарный гломерулосклероз. Прогрессирование поражения ведет к атрофии и дистрофии канальцев, интерстициальному фиброзу. При электронной микроскопии выявляются истончение, расщепление, нарушение структуры базальной мембраны. Клиническая картина разнообразна по развитию, проявлениям и течению. Выделяют 3 стадии течения нефрита: в первую стадию самочувствие ребенка не страдает, отмечается изолированный мочевой синдром, нет нарушения функции почек; вторая стадия характеризуется ухудшением самочувствия, нарастанием изменений в моче и почечной недостаточности тубулярного типа; третья стадия – терминальная - развивается к 20-30 годам, иногда раньше.
Первые признаки поражения почек при синдроме Альпорта обычно выявляют в возрасте от 3 до 10 лет. Обычно их выявляют случайно, в виде изолированного мочевого синдрома. Наиболее частым и первым признаком заболевания является гематурия различной степени выраженности. Но иногда ранним признаком заболевания бывает и протеинурия или, реже, снижение слуха. Обычно эти признаки выявляют в среднем в 6-летнем возрасте.
Гематурия при наследственном нефрите может спонтанно как появляться, так и исчезать. Очень часто она провоцируется острой респираторной вирусной инфекцией. Эритроциты в моче обычно дисморфичны, обычно обнаруживают эритроцитарные цилиндры. Протеинурии может не быть в первые годы, нередко она минимальная и имеет интермиттирующий характер. Редко отмечаются протеинурия более 2 г/сут и развитие нефротического синдрома.
Возможен наследственный нефрит с тромбоцитопенией и лейомиоматозом. Первоначально выявляют лейомиому пищевода (доброкачественная опухоль, исходящая из мышечной оболочки) с преимущественной локализацией в грудной его части. Трахеобронхиальная локализация встречается реже, но она может быть причиной летального исхода вследствие бронхоспазма. Несколько позже появляется лейомиома половых органов. Описаны случаи локализации лейомиом в области клитора, малых и больших половых губ.
У девочек заболевание чаще проявляется рецидивирующей гематурией. У мальчиков клиническое течение заболевания более тяжелое, чем у девочек. Ухудшению состояния способствуют интеркуррентные заболевания, усиление физической нагрузки, инсоляция.
Глухота чаще встречается у мальчиков, чем у девочек, развивается приблизительно к 10 годам. Снижение слуха выявляют у 74% мальчиков и у 5% девочек. Оно имеет неврогенное происхождение, выражено в различной степени, с возрастом прогрессирует от умеренного до полного. На ранних этапах снижение слуха происходит на высоких частотах, распространяясь позднее на более низкие, переходя из звукопроводящей в звуковоспринимающую тугоухость. На ранней стадии заболевания при аудиометрии выявляют невосприимчивость звуков с частотой 6-8 кГц, а впоследствии и более низких частот (4,1-2 кГц). Поражение VIII пары черепномозговых нервов или кортиева органа бывает чаще двусторонним. Ранняя тугоухость косвенно указывает на тяжесть почечного процесса. При гистологическом исследовании внутреннего уха выявляют различные изменения, среди которых чаще всего - потерю нейронов и волосяных клеток, атрофию спиральных связок, дегенерацию stria vascnlaris.
Глазные аномалии проявляются изменением полей зрения, аномалиями хрусталика и роговицы. Для синдрома Альпорта свойственны катаракта, задний лентиконус, задняя полиморфная дистрофия роговицы, псевдоотек сосочков, дистрофия сетчатки, телеангиэктазия сетчатки, нарушение цветового восприятия, колобома, страбизм, нистагм, прогрессирующий двусторонний кератоконус. Нередко выявляют нистагм и миопию. При офтальмологическом исследовании у больных часто выявляется снижение остроты зрения, передний лентиконус, пятна на сетчатке, катаракта, кератоконус.
Микроневрологическая симптоматика встречается у 90% больных с наследственным нефритом. У трети больных отмечаются симптомы вегетативной дисфункции - колебания АД, эмоциональная лабильность, головная боль, гипергидроз ладоней и стоп. Иногда определяются симптомы пирамидной недостаточности (гиперрефлексия и др.), сглаженность носогубных складок, асимметрия сухожильных рефлексов. Нарушения памяти и снижение интеллекта встречаются редко.
Для наследственного нефрита характерны признаки дисэмбриогенеза. На экскреторных урограммах иногда выявляют лоханочную эктазию, удвоенную почку, патологическую подвижность, незавершенный поворот почки.
При наследственном нефрите наблюдаются снижение уровней Т- и В-популяций лимфоцитов, IgA, склонность к повышению концентраций IgM и IgG. Снижена фагоцитарная активность. Снижение общей резистентности организма предрасполагает к пиелонефриту, гнойному отиту, частым простудным заболеваниям.
Функциональное состояние почек сохранено в стадии скрытых клинических проявлений или компенсации. В стадии субкомпенсации превалируют ренальные дисфункции по тубулярному типу с исходом в тотальную ХПН. При наследственном нефрите в биоптатах почек у детей с возрастом увеличиваются соотношение интерстиций/кора и количество склерозированных гломерул, которые являются маркерами рубцевания почек.
В ранние сроки заболевания диагностировать заболевание сложно, поскольку нет патогномоничных симптомов. Диагноз синдрома Альпорта устанавливают на основании обнаружения у ребенка нефропатии с гематурией при наличии в семье больного с аналогичной патологией и сочетании поражения почек с глухотой у самого больного или кого-то из членов семьи. Поэтому для постановки диагноза важно составлять родословную семьи больного.
Согласно данным Clifford et al. (1993), диагностическим критерием служит наличие 3 из 5 признаков, один из которых относится к почкам: 1) гематурия или смерть от ХПН в семейном анамнезе; 2) гематурия или нефротический синдром у пациента; 3) изменения гломерулярных базальных мембран (при электронной микроскопии биоптата почки); 4) снижение слуха (по данным аудиограммы); 5) врожденная патология зрения.
Для подтверждения диагноза используют биопсию почек. Для синдрома Альпорта характерны неравномерность контуров гломерулярной базальной мембраны, расслоение или сетеобразность ее плотной пластинки.
Эффективных методов патогенетической терапии наследственного нефрита нет. Лечение предусматривает организацию щадящего режима. Ограничивают физические нагрузки, не проводят профилактические прививки. Диета высококалорийная, сбалансированная, с учетом функционального состояния почек. При отсутствии признаков нарушения функции почек назначают диету с достаточным содержанием белков, жиров и углеводов. Но диета с ограничением белков, липидов, кальция и фосфора отдаляет сроки развития ХПН. Сообщено об успешном применении в комплексном лечении наследственного нефрита ингибиторов ангиотензинпревращающего фермента, которые уменьшают выраженность протеинурии и замедляют прогрессирование заболевания. Используют активаторы обмена, как пиридоксин (по 2-3 мг/кг/сут в 3 приема в течение 2-4 нед), кокарбоксилаза (по 50 мг внутримышечно через 1 сут; 10-15 инъекций), АТФ (по 1 мл внутримышечно через 1 сут; 10-15 инъекций), витамин А (по 1000 ЕД/год жизни в сутки в 1 прием; 10-14 сут), витамин Е (по 1 мг/кг/сут за 1 прием; 10-14 сут). Указанные препараты назначают курсами 2-3 раза в год. Эффективна также фитотерапия. В качестве иммуностимуляторов назначают левамизол (декарис) по 2 мг/кг/сут 2-3 раза в 1 неделю с 4-дневным перерывом. При развитии хронической почечной недостаточности проводится гемодиализ и трансплантация почки. Успех диализа и трансплантации зависит от подбора трансплантата и наличия антител к ГБМ. Антибактериальная, иммуносупрессивная и стероидная терапия показаны в пред- и посттрансплантационный периоды. Коррекцию зрения проводят с помощью очков или контактных линз. Описан положительный опыт имплантации хрусталика и оперативного лечения переднего лентиконуса.
Больные с наследственным нефритом находятся на диспансерном учете в течение всей жизни. Прогностически неблагоприятными критериями течения наследственного нефрита являются: принадлежность к мужскому полу; раннее развитие ХПН у членов семьи; протеинурия (уровень протеина более 1 г/сут); утолщение гломерулярных базальных мембран (при электронной микроскопии); неврит слухового нерва и делеция в гене COL4A5.
Острая почечная недостаточность
Острая почечная недостаточность (ОПН) – неспецифический синдром, развивающийся вследствие острой транзиторной или необратимой утраты гомеостатических функций почек, обусловленной гипоксией почечной ткани с последующим преимущественным повреждением канальцев и отеком интерстициальной ткани.
Синдром проявляется нарастающей азотемией, электролитным дисбалансом, декомпенсированным метаболическим ацидозом и нарушением способности к выделению воды. Тяжесть ОПН зависит от степени вовлечения в патологический процесс структур почечной ткани.
В зависимости от этиологии ОПН выделяют:
Преренальные причины, вызывающие нарушение почечного кровотока в связи с острой гиповолемией, артериальной гипртензией, компенсаторной централизацией кровообращения: шок травматический, инфекционный, постгеморрагический, острая дегидратация при ожогах, истощающих поносах, реже неукротимой рвоты и передозировке диуретиков, уменьшение сердечного оттока при сердечной недостаточности;
Ренальные причины, обусловливающие непосредственное повреждение нефрона: гломерулонефриты, васкулиты при диффузных заболеваниях соединительной ткани (системная красная волчанка, болезнь Шенлейна-Геноха), острый интерстициальный нефрит;
Постренальные причины, затрудняющие отток мочи: камни, закупорка кристаллами мочевой кислоты (уратная нефропатия) и сульфаниламидов, сгустками крови, опухолевой тканью, инфравезикальная обструкция нижних мочевых путей.